【休克-哈伯德方法】
拼译:Hückel-Hubbard method
此法结合休克近似和哈伯德近似,是一种简单的π电子组态相互作用理论。此理论方法注意到分子体系的拓扑性质,具有物理图象清晰和计算简化的优点,有广阔的发展应用前景。
1931年休克(E.Hückel)提出一种适用于共轭π体系的简单分子轨道理论,后来称为休克分子轨道(HMO)法。HMO法注意到分子体系的拓扑性质,具有简单而便于概括规律性的优点,其不足在于过分粗略。1963~1964年霍夫曼(R.Hoffmann)将HMO法推广为考虑全部价电子和计算重叠积分的方法,称之为推广的休克分子轨道(EHMO)法。此法较HMO法精确,但失去了HMO法中所含的分子拓扑性。1976年马生(F.A.Matsen)在多体理论中将休克近似和哈伯德近似结合起来,从而形成了一种简单的π电子组态相互作用理论,称之为休克-哈伯德方法(H-H方法)。H-H方法保留了HMO法中所含的分子拓扑性,并在一定程度上体现了π电子的组态相互作用,故有其特殊的优越性。按无自旋理论,从ρ个正交归一化轨道的所有N重积可构造无自旋矢量空间V(ρN),此空间中N电子体系的哈密顿算符H可表为: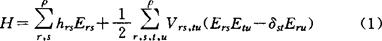
其中δst为克罗内克(Kronecker)符号,单电子积分hrs、双电子积分Vrs,tu和酉群U(ρ)的无穷小生成元(也称为基本对称算符)Ers的定义式分别是
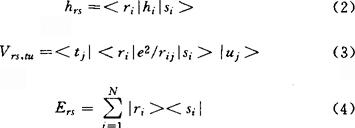
上式中hi表示单电子算符;ri表示电子i处于用r标记的轨道,但不含自旋,si、tj和uj等也有类似的意义。对hrs和Vrs,tu的处理,有各种不同的近似方法,最简单的近似处理方法就是采用休克近似。这就是将(1)式理解为共轭分子中π电子体系的哈密顿算符H的表示式,其中r、s等表示碳原子的π原子轨道(即p轨道),并令
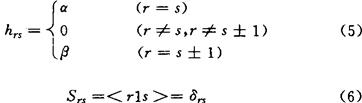
Vrs,tu=0 (7)
其中α和β为休克参数,Srs为重叠积分。可见休克近似不仅将hrs参数化和忽略π原子轨道间的重叠,而且忽略了所有的双电子积分。1976年马生提出一种改进休克近似的方法,这就是保留休克近似中的(5)和(6)式而修改(7)式。他采用哈伯德(J.Hubbard)在1963年讨论固体能带理论时所用的近似,即只考虑双电子积分项的单中心积分部分并使之参数化,故令
Vrs,tu=Iδrsδstδtu (8)
其中Ⅰ称为哈伯德电子排斥参数。在休克近似中用(8)式代替(7)式,即成为休克-哈伯德近似,由此导出的理论计算方法即是H-H方法。
马生在1976年提出H-H方法时,同时采用休克参数为反应坐标讨论了乙烯的顺一反异构、[2+2]环加成和[1、3]σ键迁移反应。计算结果表明,对自旋单重态(S0)的基态乙烯,其能量E(S0)在两个亚甲基形成的二面角Φ=90°时出现高的能峰,因而基态乙烯的顺一反异构因受到高能势垒的阻碍而难以进行。第一和第二激发态分别为自旋三重态(T1)和自旋单重态(S1),相应的能量·E(T1)和E(S1)对二面角Φ而言均不存在能垒,故激发态乙烯容易进行顺一反异构。此外,计算所得的能量曲面表明,同面一异面环加成[2s+2a]的活化能低于同面一同面环加成[2s+2s],异面[1、3]σ键迁移的活化能低于同面[1、3]σ键迁移。1983年孙家钟、封纪康等应用H-H方法对[2+2]环加成反应进行了更深入的研究。在他们的工作中,采用分子轨道构造盖尔方德(Gelfand)基并引用局部对称性进行对称性分析,计算所得的活化能值见表1。表1 [2s+2s]和[2s+2a]的活化能(eV)

表2 丁二烯电环合的非绝热跃迁几率
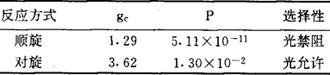
其中gc为第1激发单重态和基态的非绝热偶合项,P为此两态间的非绝热跃迁几率。上述有关周环反应的结论都与著名的伍德沃德-霍夫曼(Woodward-Hoffmann)定则相符。
1985年福克斯(M.A.Fox)和马生在H-H方法中引入电子结构图来说明π体系的电子结构。他们定义π体系的对比能量ER(K)=E(K)/(Ι-β)和价键指数x=I/(Ι-β),并将ER(K)表为x的函数。根据计算,乙烯π体系4个态的对比能量分别为: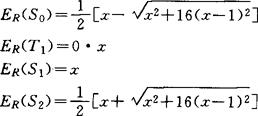
ER(K)对x所作的图称为π-CI图。用直线来代替π-CI图中的曲线,即得π体系的电子结构图,故后者是前者的简化结果。乙烯π体系的电子结构图如图1所示。图中填有休克分子轨道A、B的方格和填有碳原子2p轨道a、b的方格,分别表示乙烯π体系的分子轨道(MO)和原子轨道(AO)盖尔方德态;Sv(′S)表示乙烯基态为自旋单重态,它对分子中的对称面σV是对称的;T1(3A)表示第1个自旋三重态,它对σV是反对称的,其它的标记也有类似的意义。当x=0(I=0)时,休克-哈伯德近似还原为休克近似,这时乙烯的π电子是完全离域的,即处于休克态;当x=1(β=0)时,乙烯的π电子被驱入原来的碳原子轨道,因而是完全定域的,即处于价键态。在这两种极端情况下,各态能量大小顺序都与实际不符。实际的能量大小顺序是ER(S0)<ER(T1)<ER(S1)<ER(S2),即0<x<1。当取x=0.6时,所得乙烯π体系各态的能量数值则与实验结果基本一致。按电子结构图,π体系的电子结构既有休克态成分也有价键态成分,因而可看成是二者的混合。
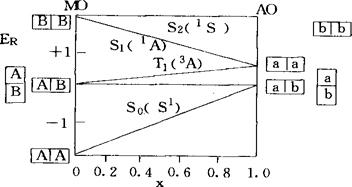
图1 乙烯π体系电子结构
H-H方法除了用于讨论共轭π体系的电子结构和协同反应的立体选择性以外,用于研究原子团簇的结构和性质也是很有希望的。事实上,EHMO和H-H方法都是休克方法的改进,前者已成为量子化学常用方法之一,后者还有待进一步研究和发展。【参考文献】:1 Hückel E Z. Physik,'l931,70:204,1932;76:6282 Hubbard J. Proc Roy Soe,London,1963,276A:2383 Matsen F A. Int J Quantum Chem,1976,10:5114 Sun Chia -chung, et al. J Phys Chem, 1983,87:34125 Fox M A.et al. J Chem Educ, 1985,62:367;4776 何福城,等.庆祝唐敖庆教授执教五十年学术论文专集.长春:吉林大学出版社,1990.517 李象远,等.高等学校化学学报,1992,13∶834(成都科技大学何福城教授撰)